希少疾患の全世界的な疾患啓発活動である 「不屈の力(Uncommon Strength)キャンペーン」の 日本語サイト開設
― 希少疾患に関わる人々を結び付け、勇気と不屈の精神を伝えるために ― ― 希少疾患の患者さんが作成したヒーローを希少疾患コミュニティで共有し、 ソーシャルメディアのプロフィール写真をアップデートすることで、 aHUS、PNH、HPP、LAL-Dに関する認知向上を世界に促す ―
キャンペーン
2016年9月28日 10:00アレクシオンファーマ合同会社(本社:東京都渋谷区)は、アレクシオン・ファーマシューティカルズ(本社:米国コネチカット州ニューヘイブン)が9月12日より開始した4つの希少疾患に関する疾患啓発活動である「不屈の力(Uncommon Strength)キャンペーン」の日本語サイト( http://www.UncommonStrength.com/ja )が開設しましたので、お知らせいたします。
このキャンペーンは、希少疾患の患者さんをはじめ希少疾患に関わる人々の不屈の精神や内に秘めた力を伝えることで希少疾患の認知を高めていこうという、アレクシオンの世界的な取り組みで、不屈の力(Uncommon Strength)のキャンペーンサイト( http://www.UncommonStrength.com )を中心に展開されます。本キャンペーンサイトには、非典型溶血性尿毒症症候群(aHUS)、発作性夜間ヘモグロビン尿症(PNH)、低ホスファターゼ症(HPP)、ライソゾーム酸性リパーゼ欠損症(LAL-D)に関する疾患情報が掲載されています。さらに同サイトではインタラクティブなソーシャルメディアを利用してコミュニティを団結させることで、これらの希少疾患コミュニティを世界的に支援します。
希少疾患は認知度が低いため、誤解や誤診されることも多く、診断に至るまでの道のりが長く困難なケースが少なくありません。実際、最初の症状が現れてから正しく診断されるまでの平均期間は約5年で、その間に病院を7回以上変えている場合もあるとされています(参考文献1)。そのため、希少疾患の患者さんとそのご家族は辛抱強く「不屈の力(Uncommon Strength)」を持ち続けることが必要です。
アレクシオンファーマ合同会社 社長のヘルマン ストレンガーは、次のように述べています。
「私たちは、希少疾患とともに生きる患者さんやそのご家族が、適時に正確な診断が受けられない、また適切な治療を受けられないなど、今なおさまざまな困難に直面していること、そしてその困難に立ち向かうために内に秘めた力と忍耐力が必要であることを十分に認識しています。
私たちアレクシオンにとって希少疾患の患者さんこそが真のヒーローであり、この不屈の力(Uncommon Strength)キャンペーンによって多くの患者さんやそのご家族が情報を共有し、疾患に対する認知が高まることが私たちの願いです。」
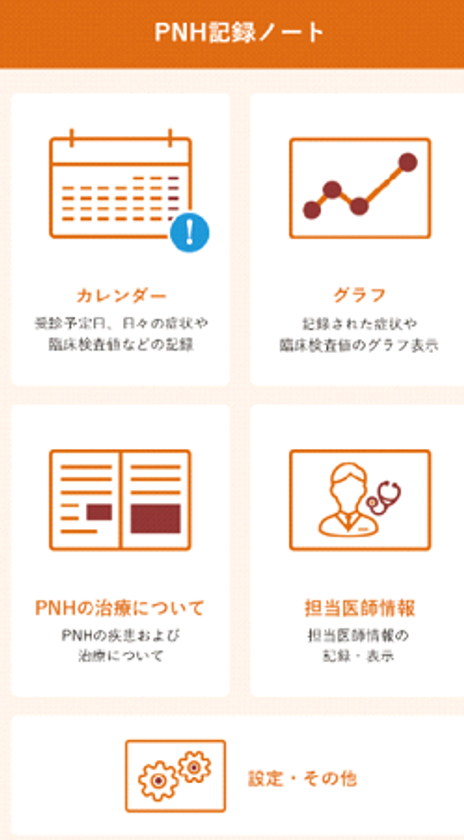
希少疾患の患者さんと、そのご家族、介護者の方、大切な仲間といった希少疾患に関わる皆さんは以下のキャンペーンサイトを是非ご覧ください。
http://www.UncommonStrength.com/ja では:
・あなたならではの勇気や力、忍耐力を示すために、aHUS、PNH、HPP、LAL-Dなどの希少疾患のヒーローを各自で作成して、希少疾患コミュニティを支援してください。
・各自が作成したヒーローをソーシャルメディア等で共有して、希少疾患についての対話を広め、世界中の人々が更に興味を持って啓発活動に参加するよう促してください。
・今回のキャンペーンのために作成したフォトフィルターを用いて個人のFacebookやTwitterのプロフィール写真をアップデートして、aHUS、PNH、HPP、LAL-Dなどの希少疾患について多くの人に伝えてください。
「不屈の力(Uncommon Strength)キャンペーン」の期間中には、以下が予定されています:
・2016年9月24日
― aHUS啓発デー(aHUS Awareness Day)
・2016年9月26日~10月2日
― 米国PNH啓発週間(PNH Awareness Week in the United States)
・2016年10月13日
― 世界血栓症デー(World Thrombosis Day)
・2016年10月24日~30日
― HPP啓発週間(HPP Awareness Week)
・2016年10月
― 肝臓啓発月間(Liver Awareness Month)
aHUS、PNH、HPP、LAL-Dは、患者数が100万人あたり20人に満たない疾患と定義されている超希少疾患です(参考文献2)。「不屈の力(Uncommon Strength)キャンペーン」は疾患啓発を目的としており、希少疾患のコミュニティを活性化してつながりを生み出すことで希少疾患の認知向上を世界的に訴えかけるとともに、希少疾患の患者さんやそのご家族が必要としている情報を確実に入手できる場を提供します。
以下の患者団体・患者支援団体が「不屈の力(Uncommon Strength)キャンペーン」の支援サポーターで、キャンペーンサイトから各団体にアクセスすることが可能です。日本からは、NPO法人PNH倶楽部と低フォスファターゼ症の会(HPPSA-J)が、支援サポーターとして本活動に賛同いただいています。
<aHUS>
○aHUS Alliance
○aHUS UK
○Atypical HUS Foundation
○Selbsthilfe fur seltene komplementbedingte Erkrankungen MPGN und aHUS e.V.
<PNH>
○NPO法人PNH倶楽部
○Aplastic Anemia and MDS International Foundation(AAMDSIF)
○Aplastic Anemia & Myelodysplasia Association of Canada(AAMAC)
○Canadian Association of PNH Patients
<HPP>
○低フォスファターゼ症の会(HPPSA-J)
○Children Living with Inherited Metabolic Diseases (CLIMB)
○Hypophosphatasie Deutschland e.V.
○International Coalition of Organizations Supporting Endocrine Patients(ICOSEP)
○Soft Bones
○The MAGIC Foundation
<LAL-D>
○LAL Solace
○AE LALD Spain
<Rare Disease>
○Federacion Colombiana de Enfermedades Raras(FECOER)
○Federacion Mexicana de Enfermedades Raras(FEMEXER)
本キャンペーンに参加してaHUS、PNH、HPP、LAL-Dの各疾患の認知向上を支援する方法についてさらに詳しく知りたい方は http://www.UncommonStrength.com/ja をご覧ください。
■非典型溶血性尿毒症症候群(aHUS)について
aHUSは、極めて希少で生命を脅かす慢性疾患であり、1つ以上の補体制御遺伝子の遺伝子異常が生涯にわたる慢性的で制御不能な補体の活性化を引き起こし、その結果、全身の微小血管に血栓を形成する補体介在性の血栓性微小血管障害症(TMA)を発症します(参考文献3、4)。aHUSにおける慢性的な制御不能な補体の活性化は生涯にわたるTMAリスクの原因となり、結果として腎臓、脳、心臓およびその他の重要な臓器の突然の破壊的かつ致死的な損傷や早期死亡に至ります(参考文献3、5)。全aHUS患者さんの79%が、血漿交換または血漿輸注(PE/PI)を実施しても、診断後3年以内に死亡、または腎透析を必要とするか、または永続的な腎障害に至ります(参考文献6)。さらに、PE/PIを行っても、33~40%の患者さんが初回のaHUS発作によって死亡するか、または末期腎不全へと進行します(参考文献6、7)。腎移植を受けたaHUS患者さんの大半は後に全身性TMAを発症し、これらのTMA患者さんにおける移植失敗率は90%に達します(参考文献8)。
小児と成人のいずれの年代においてもaHUSに罹病します。また、補体介在性TMAは血小板数の減少(血小板減少症)と赤血球破壊(溶血)をもたらします。少なくとも10種類の補体制御遺伝子において遺伝子変異が同定されているものの、aHUSの確定診断を受けた患者さんの30~50%で遺伝子変異は同定されていません(参考文献8)。
■発作性夜間ヘモグロビン尿症(PNH)について
PNHは、正常な免疫系の構成要素である補体の機能のコントロールができなくなり、溶血(赤血球破壊)を引き起こす極めて稀な血液疾患です。PNHはどの年齢の人もかかる病気ですが、平均発症年齢は30代初期です(参考文献9)。患者さん全体の約10%では、21歳以下で初めて症状が現れます(参考文献10)。PNHは前触れもなく進行し、全ての人種、背景または年齢の男女に発症する可能性があります。PHNは気づかないうちに進行し、およそ1~10年以上診断が遅れることがあります(参考文献11)。治療薬が発売される前は、推定でPNH患者さんの約3分の1は診断後5年以上生存できませんでした(参考文献9)。PNHは再生不良性貧血(AA)や骨髄異形成症候群(MDS)などの骨髄不全例に一般的に多く認められてきました(参考文献12-14)。原因不明の血栓を呈する患者さんは、PNHが根本原因の可能性があります(参考文献9)。
■低ホスファターゼ症(HPP)について
HPPは、アルカリホスファターゼ(ALP)活性の低下と骨石灰化不全を特徴とする、極めて稀な慢性進行性の遺伝性代謝性疾患で骨の変形等の骨格異常の他、重度の筋力低下、けいれん発作、疼痛といった全身性の合併症を生じ、乳児では呼吸不全によって早期死亡に至ることもあります(参考文献15-19)。
HPPは組織非特異型アルカリホスファターゼ(TNSALP)と呼ばれる酵素の遺伝子コードの変異が原因で発症します(参考文献15、16)。HPPにおける遺伝子異常は、あらゆる年齢層の患者さんに影響を及ぼします(参考文献15)。HPPは、乳児、小児、成人に症状が現れる可能性があり、疾患発症時の年齢によって分類されます。
HPPは、年齢を問わず、患者さんに深刻な影響を及ぼす可能性があります(参考文献15)。自然経過観察試験によると、生後6ヵ月未満にHPPを発症した乳児の死亡率は高く、5歳時の全死亡率は73%と報告されています(参考文献20)。このような患者さんの死亡原因は主として呼吸不全です(参考文献15、19、21)。発症年齢にかかわらず、長期的な臨床経過には、成長と発達への影響、日常生活動作(立つ、階段の上り下り等)への支障、再発性および治癒しない骨折、重度の筋力低下、重度の疼痛、ならびに車椅子、歩行器および杖などの歩行補助器が必要とされる運動機能の障害などがあります(参考文献15、18)。
■ライソゾーム酸性リパーゼ欠損症(LAL-D)について
LAL-Dは、生命を脅かす遺伝性疾患で、進行性の多臓器障害により、早期に死亡に至る可能性があります(参考文献22)。LAL-D患者さんでは、遺伝子変異によりLAL酵素活性が低下または欠損するため、重要な臓器や血管、その他の組織にコレステロールエステルやトリグリセリドが著しく蓄積します。その結果として組織の線維化、肝硬変、肝不全、アテローム性動脈硬化症が進み、心疾患やその他の深刻な疾患を含む、進行性の多臓器障害をもたらします(参考文献22、23)。
LAL-Dはあらゆる年齢の患者さんに影響を及ぼし、乳児期から成人期にわたって臨床症状が現れ、突発的かつ予想不可能な臨床的合併症が現れる可能性があります。乳児の患者さんでは、重篤な成長障害、肝線維症、肝硬変を合併し、生存期間の中央値は3.7ヵ月です(参考文献24)。乳児期を超えてから発症したLAL-D患者さんを対象とした観察研究によると、小児および成人のLAL-D患者さんの約50%が3年以内に組織の線維化、肝硬変、肝移植に至っています(参考文献25)。LAL-Dの発症年齢は5.8歳(中央値)であり、簡単な血液検査で診断できます(参考文献26、27)。
■アレクシオンファーマ合同会社について
アレクシオンファーマ合同会社は、アレクシオン・ファーマシューティカルズ(米国コネチカット州ニューヘイブン)の日本法人です。アレクシオンは、重篤な希少疾患を抱える患者さんの生活を一変させる治療薬の開発と提供に注力するグローバルなバイオ製薬企業です。アレクシオンは補体阻害におけるグローバルリーダーであり、生命を脅かす2つの超希少疾患である発作性夜間ヘモグロビン尿症(PNH)および非典型溶血性尿毒症症候群(aHUS)の治療薬として初めてかつ唯一承認されている補体阻害薬を開発し、製造販売しています。また、アレクシオンの代謝性フランチャイズは、低ホスファターゼ症(HPP)とライソゾーム酸性リパーゼ欠損症(LAL-D)といった生命を脅かす超希少疾患の患者さんに対する2つの非常に革新的な酵素補充療法を有しています。さらに、アレクシオンは、複数の治療領域にわたる極めて革新的な製品候補を擁し、バイオテクノロジー業界において最も強固な希少疾患パイプラインを進展させています。本プレスリリースとアレクシオンファーマ合同会社に関する詳細については http://alexionpharma.jp をご覧ください。
■参考文献
1. Engel PA, Gabal S, Broback M, Boice N. Physician and patient percepts regarding physician training in rare diseases: the need for stronger educational initiatives for physicians. Journal of Rare Diseases. 2013;1(2):1-15.
2. REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32000R0141&qid=1421232987002&from=EN.
3. Benz K, Amann K. Thrombotic microangiopathy: new insights. Curr Opin Nephrol Hypertens. 2010;19(3):242-7.
4. Ariceta G, Besbas N, Johnson S, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24:687-96.
5.Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70(1):16-23.
6. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676-87.
7. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844-59.
8. Bresin E, Daina E, Noris M, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88-99.
9. Socie G, Mary JY, de Gramont A., Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet 1996 Aug 31;348(9027):573-7.
10. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-3709.
11. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258.
12. Wang H, Chuhjo T, Yasue S, Omine M, Naka S. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome. Blood. 2002;100 (12):3897-3902.
13. Iwanga M, Furukawa K, Amenomori T, et al. Paroxysmal nocturnal haemoglobinuria clones in patients with myelodysplastic syndromes. Br J Haematol. 1998;102(2):465-474.
14. Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clones. Br J Haematol. 2001;115:1015-1022.
15. Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013; 10(2):380-388.
16. Whyte MP. Hypophosphatasia: nature’s window on alkaline phosphatase function in humans. In: Bilezikian JP, Raisz LG, Martin TJ, eds. Principles of Bone Biology. Vol 1. 3rd ed. San Diego, CA: Academic Press; 2008:1573-1598.
17. Whyte MP, Greenberg CR, Salman N, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med.2012; 366(10):904-913.
18. Seshia SS, Derbyshire G, Haworth JC, Hoogstraten J. Myopathy with hypophosphatasia. Arch Dis Child. 1990; 65(1):130-131.
19. Baumgartner-Sigl S, Haberlandt E, Mumm S, et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007; 40(6):1655-1661.
20. Whyte MP, Leung E, Wilcox W, et al. Hypophosphatasia: a retrospective natural history study of the severe perinatal and infantile forms. Poster presented at the 2014 Pediatric Academic Societies and Asian Society for Pediatric Research Joint Meeting, Vancouver, B.C., Canada, May 5, 2014. Abstract 752416.
21. Whyte MP, Rockman-Greenberg C, Hofmann C, et al. Improved survival with asfotase alfa treatment in pediatric patients with hypophosphatasia at high risk of death. Poster presented at the American Society for Bone and Mineral Research (ASBMR) 2014 Annual Meeting, Houston, September 14, 2014. Abstract 1097.
22. Bernstein DL, et al. Chloesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230-43.
23. Reiner Z, et al. Lysosomal acid lipase deficiency ? an under-recognized cause of dyslipidemia and liver dysfunction. Atherosclerosis. 2014;235:21-30.
24. Jones SA et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 27 August 2015. doi:10.1038/gim.2015.108.
25. Data on file, Alexion.
26. Burton et al. Clinical Features of Lysosomal Acid Lipase Deficiency - a Longitudinal Assessment of 48 Children and Adults. Journal of Pediatric Gastroenterology & Nutrition. 2015.
27. Hamilton J, et al. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413:1207-10. doi:10.1016/j.cca.2012.03.019.
このキャンペーンは、希少疾患の患者さんをはじめ希少疾患に関わる人々の不屈の精神や内に秘めた力を伝えることで希少疾患の認知を高めていこうという、アレクシオンの世界的な取り組みで、不屈の力(Uncommon Strength)のキャンペーンサイト( http://www.UncommonStrength.com )を中心に展開されます。本キャンペーンサイトには、非典型溶血性尿毒症症候群(aHUS)、発作性夜間ヘモグロビン尿症(PNH)、低ホスファターゼ症(HPP)、ライソゾーム酸性リパーゼ欠損症(LAL-D)に関する疾患情報が掲載されています。さらに同サイトではインタラクティブなソーシャルメディアを利用してコミュニティを団結させることで、これらの希少疾患コミュニティを世界的に支援します。
希少疾患は認知度が低いため、誤解や誤診されることも多く、診断に至るまでの道のりが長く困難なケースが少なくありません。実際、最初の症状が現れてから正しく診断されるまでの平均期間は約5年で、その間に病院を7回以上変えている場合もあるとされています(参考文献1)。そのため、希少疾患の患者さんとそのご家族は辛抱強く「不屈の力(Uncommon Strength)」を持ち続けることが必要です。
アレクシオンファーマ合同会社 社長のヘルマン ストレンガーは、次のように述べています。
「私たちは、希少疾患とともに生きる患者さんやそのご家族が、適時に正確な診断が受けられない、また適切な治療を受けられないなど、今なおさまざまな困難に直面していること、そしてその困難に立ち向かうために内に秘めた力と忍耐力が必要であることを十分に認識しています。
私たちアレクシオンにとって希少疾患の患者さんこそが真のヒーローであり、この不屈の力(Uncommon Strength)キャンペーンによって多くの患者さんやそのご家族が情報を共有し、疾患に対する認知が高まることが私たちの願いです。」
希少疾患の患者さんと、そのご家族、介護者の方、大切な仲間といった希少疾患に関わる皆さんは以下のキャンペーンサイトを是非ご覧ください。
http://www.UncommonStrength.com/ja では:
・あなたならではの勇気や力、忍耐力を示すために、aHUS、PNH、HPP、LAL-Dなどの希少疾患のヒーローを各自で作成して、希少疾患コミュニティを支援してください。
・各自が作成したヒーローをソーシャルメディア等で共有して、希少疾患についての対話を広め、世界中の人々が更に興味を持って啓発活動に参加するよう促してください。
・今回のキャンペーンのために作成したフォトフィルターを用いて個人のFacebookやTwitterのプロフィール写真をアップデートして、aHUS、PNH、HPP、LAL-Dなどの希少疾患について多くの人に伝えてください。
「不屈の力(Uncommon Strength)キャンペーン」の期間中には、以下が予定されています:
・2016年9月24日
― aHUS啓発デー(aHUS Awareness Day)
・2016年9月26日~10月2日
― 米国PNH啓発週間(PNH Awareness Week in the United States)
・2016年10月13日
― 世界血栓症デー(World Thrombosis Day)
・2016年10月24日~30日
― HPP啓発週間(HPP Awareness Week)
・2016年10月
― 肝臓啓発月間(Liver Awareness Month)
aHUS、PNH、HPP、LAL-Dは、患者数が100万人あたり20人に満たない疾患と定義されている超希少疾患です(参考文献2)。「不屈の力(Uncommon Strength)キャンペーン」は疾患啓発を目的としており、希少疾患のコミュニティを活性化してつながりを生み出すことで希少疾患の認知向上を世界的に訴えかけるとともに、希少疾患の患者さんやそのご家族が必要としている情報を確実に入手できる場を提供します。
以下の患者団体・患者支援団体が「不屈の力(Uncommon Strength)キャンペーン」の支援サポーターで、キャンペーンサイトから各団体にアクセスすることが可能です。日本からは、NPO法人PNH倶楽部と低フォスファターゼ症の会(HPPSA-J)が、支援サポーターとして本活動に賛同いただいています。
<aHUS>
○aHUS Alliance
○aHUS UK
○Atypical HUS Foundation
○Selbsthilfe fur seltene komplementbedingte Erkrankungen MPGN und aHUS e.V.
<PNH>
○NPO法人PNH倶楽部
○Aplastic Anemia and MDS International Foundation(AAMDSIF)
○Aplastic Anemia & Myelodysplasia Association of Canada(AAMAC)
○Canadian Association of PNH Patients
<HPP>
○低フォスファターゼ症の会(HPPSA-J)
○Children Living with Inherited Metabolic Diseases (CLIMB)
○Hypophosphatasie Deutschland e.V.
○International Coalition of Organizations Supporting Endocrine Patients(ICOSEP)
○Soft Bones
○The MAGIC Foundation
<LAL-D>
○LAL Solace
○AE LALD Spain
<Rare Disease>
○Federacion Colombiana de Enfermedades Raras(FECOER)
○Federacion Mexicana de Enfermedades Raras(FEMEXER)
本キャンペーンに参加してaHUS、PNH、HPP、LAL-Dの各疾患の認知向上を支援する方法についてさらに詳しく知りたい方は http://www.UncommonStrength.com/ja をご覧ください。
■非典型溶血性尿毒症症候群(aHUS)について
aHUSは、極めて希少で生命を脅かす慢性疾患であり、1つ以上の補体制御遺伝子の遺伝子異常が生涯にわたる慢性的で制御不能な補体の活性化を引き起こし、その結果、全身の微小血管に血栓を形成する補体介在性の血栓性微小血管障害症(TMA)を発症します(参考文献3、4)。aHUSにおける慢性的な制御不能な補体の活性化は生涯にわたるTMAリスクの原因となり、結果として腎臓、脳、心臓およびその他の重要な臓器の突然の破壊的かつ致死的な損傷や早期死亡に至ります(参考文献3、5)。全aHUS患者さんの79%が、血漿交換または血漿輸注(PE/PI)を実施しても、診断後3年以内に死亡、または腎透析を必要とするか、または永続的な腎障害に至ります(参考文献6)。さらに、PE/PIを行っても、33~40%の患者さんが初回のaHUS発作によって死亡するか、または末期腎不全へと進行します(参考文献6、7)。腎移植を受けたaHUS患者さんの大半は後に全身性TMAを発症し、これらのTMA患者さんにおける移植失敗率は90%に達します(参考文献8)。
小児と成人のいずれの年代においてもaHUSに罹病します。また、補体介在性TMAは血小板数の減少(血小板減少症)と赤血球破壊(溶血)をもたらします。少なくとも10種類の補体制御遺伝子において遺伝子変異が同定されているものの、aHUSの確定診断を受けた患者さんの30~50%で遺伝子変異は同定されていません(参考文献8)。
■発作性夜間ヘモグロビン尿症(PNH)について
PNHは、正常な免疫系の構成要素である補体の機能のコントロールができなくなり、溶血(赤血球破壊)を引き起こす極めて稀な血液疾患です。PNHはどの年齢の人もかかる病気ですが、平均発症年齢は30代初期です(参考文献9)。患者さん全体の約10%では、21歳以下で初めて症状が現れます(参考文献10)。PNHは前触れもなく進行し、全ての人種、背景または年齢の男女に発症する可能性があります。PHNは気づかないうちに進行し、およそ1~10年以上診断が遅れることがあります(参考文献11)。治療薬が発売される前は、推定でPNH患者さんの約3分の1は診断後5年以上生存できませんでした(参考文献9)。PNHは再生不良性貧血(AA)や骨髄異形成症候群(MDS)などの骨髄不全例に一般的に多く認められてきました(参考文献12-14)。原因不明の血栓を呈する患者さんは、PNHが根本原因の可能性があります(参考文献9)。
■低ホスファターゼ症(HPP)について
HPPは、アルカリホスファターゼ(ALP)活性の低下と骨石灰化不全を特徴とする、極めて稀な慢性進行性の遺伝性代謝性疾患で骨の変形等の骨格異常の他、重度の筋力低下、けいれん発作、疼痛といった全身性の合併症を生じ、乳児では呼吸不全によって早期死亡に至ることもあります(参考文献15-19)。
HPPは組織非特異型アルカリホスファターゼ(TNSALP)と呼ばれる酵素の遺伝子コードの変異が原因で発症します(参考文献15、16)。HPPにおける遺伝子異常は、あらゆる年齢層の患者さんに影響を及ぼします(参考文献15)。HPPは、乳児、小児、成人に症状が現れる可能性があり、疾患発症時の年齢によって分類されます。
HPPは、年齢を問わず、患者さんに深刻な影響を及ぼす可能性があります(参考文献15)。自然経過観察試験によると、生後6ヵ月未満にHPPを発症した乳児の死亡率は高く、5歳時の全死亡率は73%と報告されています(参考文献20)。このような患者さんの死亡原因は主として呼吸不全です(参考文献15、19、21)。発症年齢にかかわらず、長期的な臨床経過には、成長と発達への影響、日常生活動作(立つ、階段の上り下り等)への支障、再発性および治癒しない骨折、重度の筋力低下、重度の疼痛、ならびに車椅子、歩行器および杖などの歩行補助器が必要とされる運動機能の障害などがあります(参考文献15、18)。
■ライソゾーム酸性リパーゼ欠損症(LAL-D)について
LAL-Dは、生命を脅かす遺伝性疾患で、進行性の多臓器障害により、早期に死亡に至る可能性があります(参考文献22)。LAL-D患者さんでは、遺伝子変異によりLAL酵素活性が低下または欠損するため、重要な臓器や血管、その他の組織にコレステロールエステルやトリグリセリドが著しく蓄積します。その結果として組織の線維化、肝硬変、肝不全、アテローム性動脈硬化症が進み、心疾患やその他の深刻な疾患を含む、進行性の多臓器障害をもたらします(参考文献22、23)。
LAL-Dはあらゆる年齢の患者さんに影響を及ぼし、乳児期から成人期にわたって臨床症状が現れ、突発的かつ予想不可能な臨床的合併症が現れる可能性があります。乳児の患者さんでは、重篤な成長障害、肝線維症、肝硬変を合併し、生存期間の中央値は3.7ヵ月です(参考文献24)。乳児期を超えてから発症したLAL-D患者さんを対象とした観察研究によると、小児および成人のLAL-D患者さんの約50%が3年以内に組織の線維化、肝硬変、肝移植に至っています(参考文献25)。LAL-Dの発症年齢は5.8歳(中央値)であり、簡単な血液検査で診断できます(参考文献26、27)。
■アレクシオンファーマ合同会社について
アレクシオンファーマ合同会社は、アレクシオン・ファーマシューティカルズ(米国コネチカット州ニューヘイブン)の日本法人です。アレクシオンは、重篤な希少疾患を抱える患者さんの生活を一変させる治療薬の開発と提供に注力するグローバルなバイオ製薬企業です。アレクシオンは補体阻害におけるグローバルリーダーであり、生命を脅かす2つの超希少疾患である発作性夜間ヘモグロビン尿症(PNH)および非典型溶血性尿毒症症候群(aHUS)の治療薬として初めてかつ唯一承認されている補体阻害薬を開発し、製造販売しています。また、アレクシオンの代謝性フランチャイズは、低ホスファターゼ症(HPP)とライソゾーム酸性リパーゼ欠損症(LAL-D)といった生命を脅かす超希少疾患の患者さんに対する2つの非常に革新的な酵素補充療法を有しています。さらに、アレクシオンは、複数の治療領域にわたる極めて革新的な製品候補を擁し、バイオテクノロジー業界において最も強固な希少疾患パイプラインを進展させています。本プレスリリースとアレクシオンファーマ合同会社に関する詳細については http://alexionpharma.jp をご覧ください。
■参考文献
1. Engel PA, Gabal S, Broback M, Boice N. Physician and patient percepts regarding physician training in rare diseases: the need for stronger educational initiatives for physicians. Journal of Rare Diseases. 2013;1(2):1-15.
2. REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32000R0141&qid=1421232987002&from=EN.
3. Benz K, Amann K. Thrombotic microangiopathy: new insights. Curr Opin Nephrol Hypertens. 2010;19(3):242-7.
4. Ariceta G, Besbas N, Johnson S, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24:687-96.
5.Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70(1):16-23.
6. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676-87.
7. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844-59.
8. Bresin E, Daina E, Noris M, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88-99.
9. Socie G, Mary JY, de Gramont A., Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet 1996 Aug 31;348(9027):573-7.
10. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-3709.
11. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258.
12. Wang H, Chuhjo T, Yasue S, Omine M, Naka S. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome. Blood. 2002;100 (12):3897-3902.
13. Iwanga M, Furukawa K, Amenomori T, et al. Paroxysmal nocturnal haemoglobinuria clones in patients with myelodysplastic syndromes. Br J Haematol. 1998;102(2):465-474.
14. Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clones. Br J Haematol. 2001;115:1015-1022.
15. Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013; 10(2):380-388.
16. Whyte MP. Hypophosphatasia: nature’s window on alkaline phosphatase function in humans. In: Bilezikian JP, Raisz LG, Martin TJ, eds. Principles of Bone Biology. Vol 1. 3rd ed. San Diego, CA: Academic Press; 2008:1573-1598.
17. Whyte MP, Greenberg CR, Salman N, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med.2012; 366(10):904-913.
18. Seshia SS, Derbyshire G, Haworth JC, Hoogstraten J. Myopathy with hypophosphatasia. Arch Dis Child. 1990; 65(1):130-131.
19. Baumgartner-Sigl S, Haberlandt E, Mumm S, et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007; 40(6):1655-1661.
20. Whyte MP, Leung E, Wilcox W, et al. Hypophosphatasia: a retrospective natural history study of the severe perinatal and infantile forms. Poster presented at the 2014 Pediatric Academic Societies and Asian Society for Pediatric Research Joint Meeting, Vancouver, B.C., Canada, May 5, 2014. Abstract 752416.
21. Whyte MP, Rockman-Greenberg C, Hofmann C, et al. Improved survival with asfotase alfa treatment in pediatric patients with hypophosphatasia at high risk of death. Poster presented at the American Society for Bone and Mineral Research (ASBMR) 2014 Annual Meeting, Houston, September 14, 2014. Abstract 1097.
22. Bernstein DL, et al. Chloesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230-43.
23. Reiner Z, et al. Lysosomal acid lipase deficiency ? an under-recognized cause of dyslipidemia and liver dysfunction. Atherosclerosis. 2014;235:21-30.
24. Jones SA et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 27 August 2015. doi:10.1038/gim.2015.108.
25. Data on file, Alexion.
26. Burton et al. Clinical Features of Lysosomal Acid Lipase Deficiency - a Longitudinal Assessment of 48 Children and Adults. Journal of Pediatric Gastroenterology & Nutrition. 2015.
27. Hamilton J, et al. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413:1207-10. doi:10.1016/j.cca.2012.03.019.